Overview
Germline mutation rates are important in genetics, genomics and evolutionary biology. It is long known that mutation rates vary substantially across the genome, but existing methods can only obtain very rough estimates of local mutation rates and are difficult to be applied to non-model species.
MuRaL (short for Mutation Rate Learner), is a generalizable deep learning framework for mutation rate estimation.
Framework Components
Module |
Functionality |
|---|---|
MuRaL-snv |
Estimates single-nucleotide variant (SNV) rates |
MuRaL-indel |
Estimates small insertion/deletion (INDEL) rates (1-50bp) |
Key advantages over conventional methods:
Superior prediction accuracy across genomic scales
Genome-wide rate mapping from moderate samples (~100 individuals)
Transfer learning capability reduces data requirements
Broad species applicability with population polymorphism data
Output Demonstration
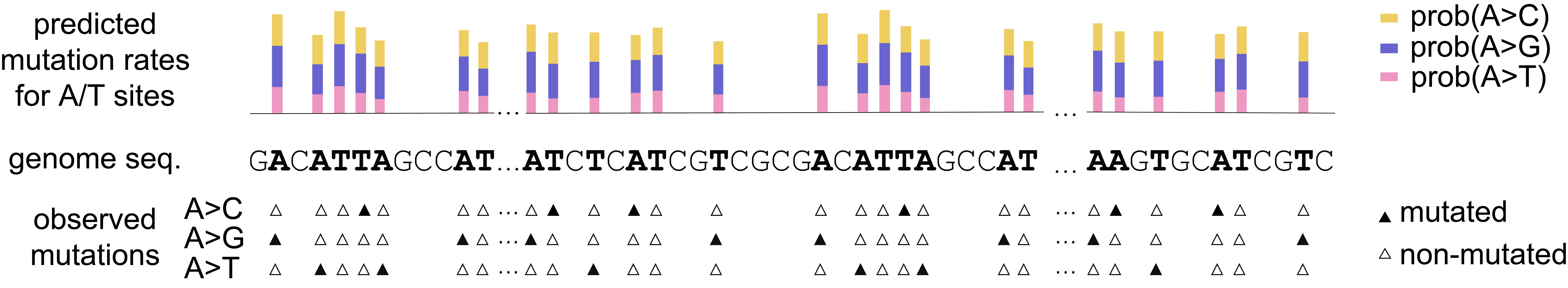
SNV Rate Prediction (A. thaliana)
Below is an example showing that MuRaL-snv predicted each A/T sites mutation rate
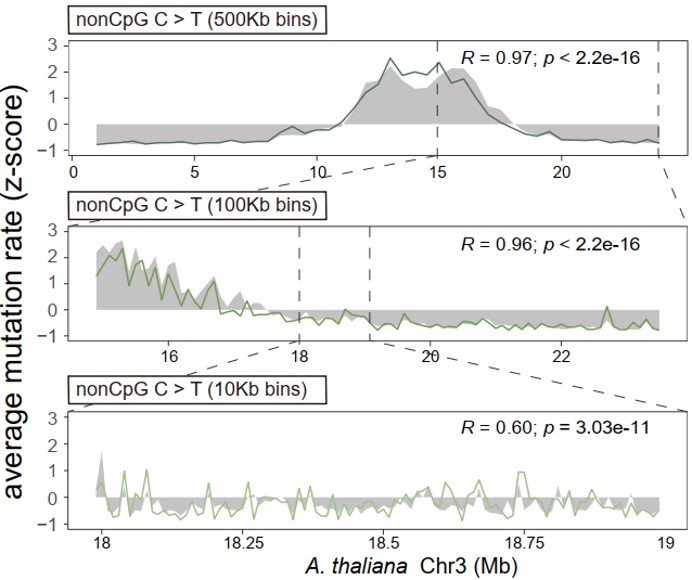
Below is an example showing that MuRaL-snv predicted rates (colored lines) are highly correlated with observed mutation rates (grey shades) at different scales on Chr3 of A. thaliana.
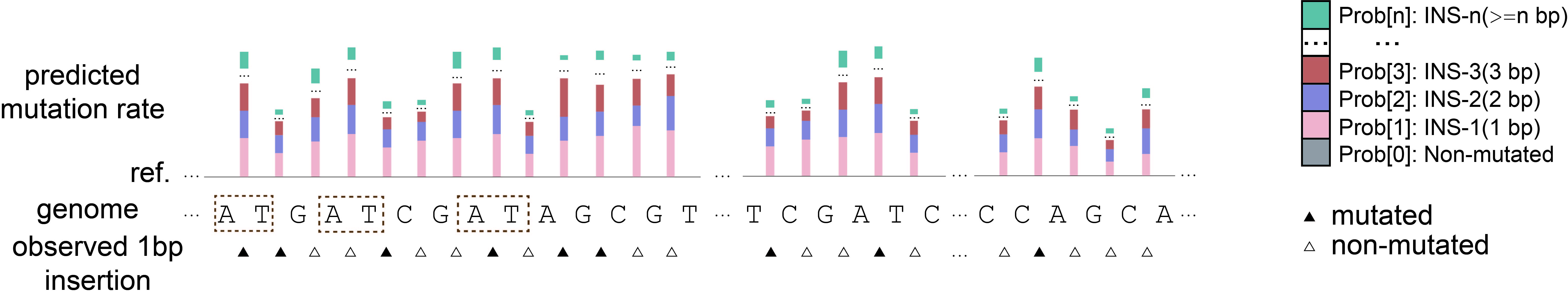
INDEL Rate Prediction (H. sapiens)
Below is an example showing that MuRaL-indel predicted each one-bp insertion mutation rate
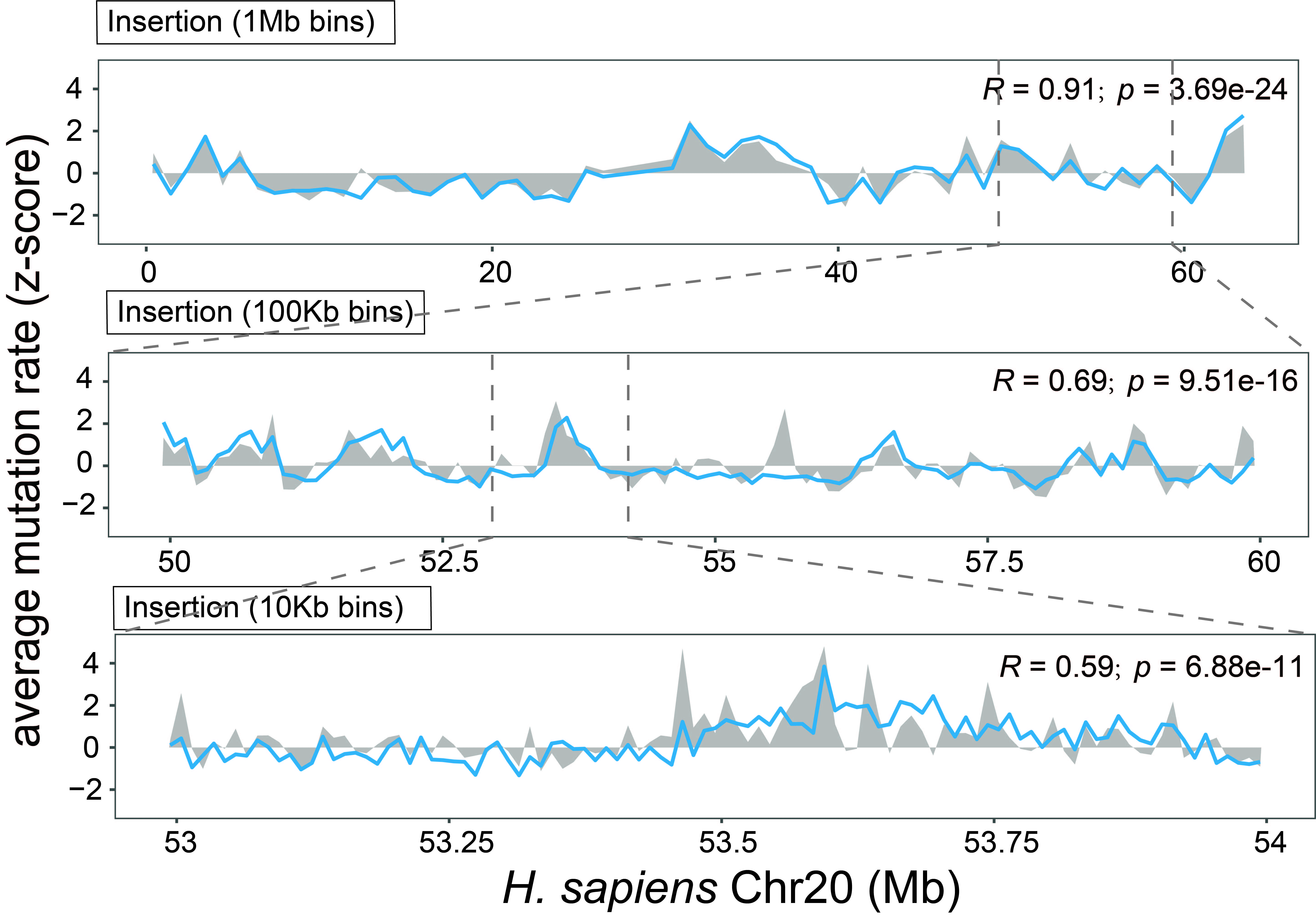
Below is an example showing that MuRaL-indel predicted rates (colored lines) are highly correlated with observed mutation rates (grey shades) at different scales on Chr20 of H. sapiens.
Model Architecture
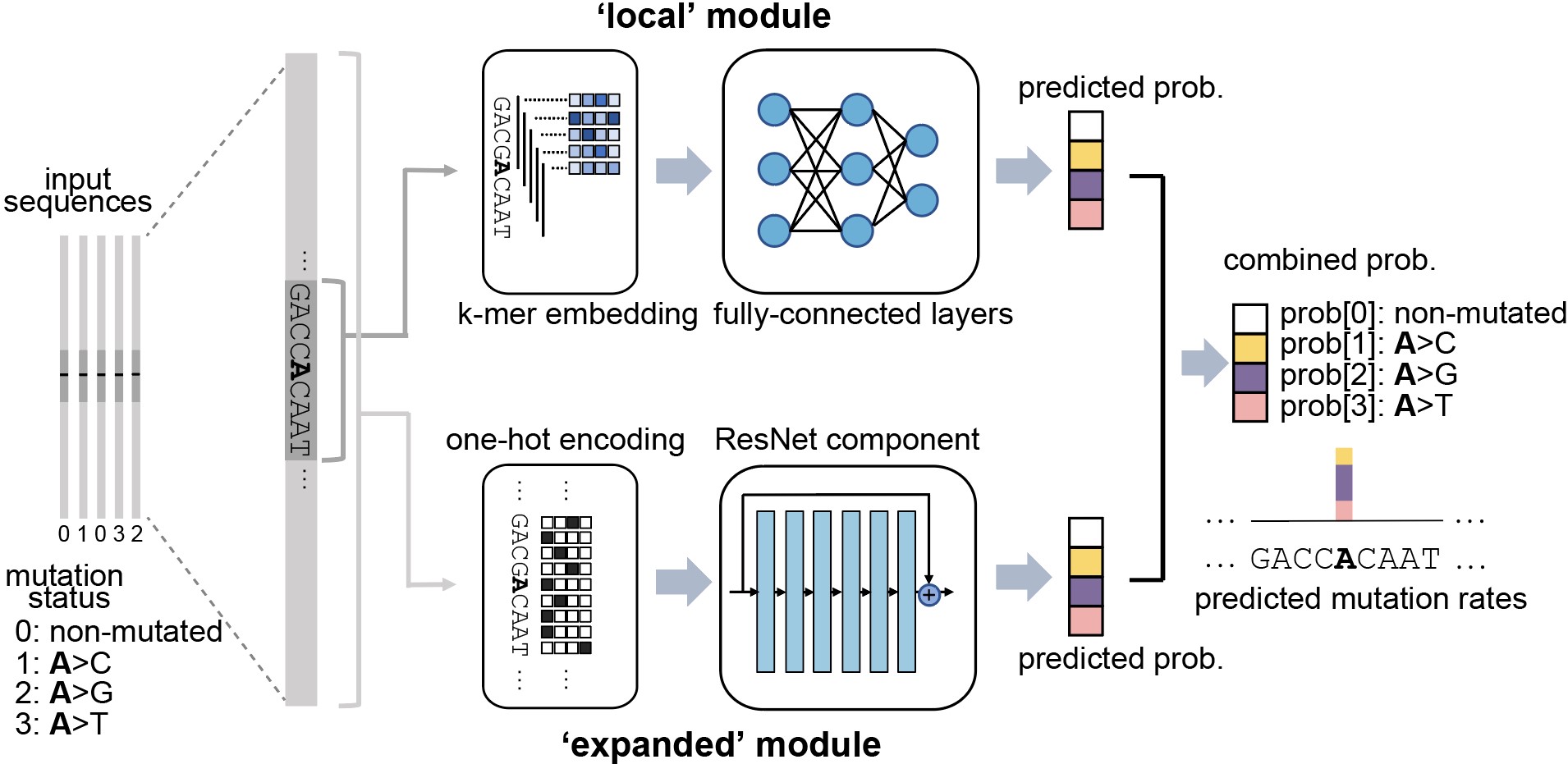
MuRaL-snv Architecture
The MuRaL-snv network architecture has two main modules (shown below), one is for learning signals from local genomic regions (e.g. 10bp on each side of the focal nucleotide), the other for learning signals from expanded regions (e.g. 1Kb on each side of the focal nucleotide).
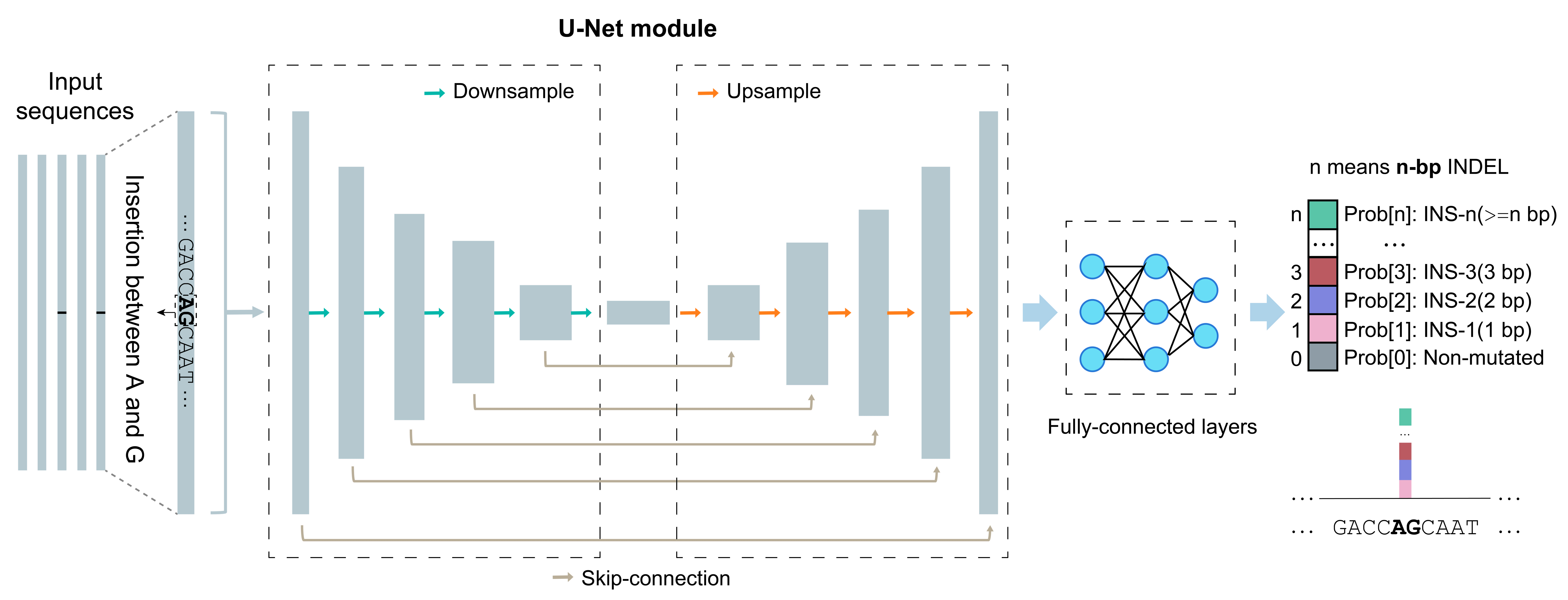
MuRaL-indel Architecture
The MuRaL-indel network architecture employs a U-Net based CNN structure (shown below) that processes long sequence contexts around potential INDEL breakpoints. The model takes one-hot encoded DNA sequences as input and outputs probabilities for different INDEL lengths (including no mutation). The U-Net consists of symmetrical downsampling and upsampling paths connected through skip connections, with residual blocks and convolutional layers at each scale to extract hierarchical features.
Install
Using Conda
MuRaL depends on several other packages. We recommend using Miniconda (version 3 or newer) to create a conda environment for installation.
Prerequisites
Install Miniconda (version 3+)
Clone/download MuRaL source code and navigate to ‘MuRaL-xxx/’ directory
System Requirements
GPU recommended for training (significantly faster)
CPU sufficient for most prediction tasks
Environment files provided:
environment.yml(GPU version)environment_cpu.yml(CPU-only version)
Installation Process
Create conda environment (may take >30 minutes depending on your internet):
# For GPU systems
conda env create -n mural -f environment.yml
# If interrupted, update instead:
conda env update -n mural -f environment.yml --prune
# For CPU-only systems
conda env create -n mural -f environment_cpu.yml
Activate environment and install MuRaL:
conda activate mural
pip install .
Verify installation:
mural_snv train -v # Should display version number
Using Singularity
Singularity is a popular container platform for scientific research. We
also built Singularity images for specific versions, which can be found
at this OSF repo. You can just download the
Singularity image mural_vx.x.x.sif from the OSF repo and don’t need
to install the dependencies of MuRaL. Once Singularity is installed in
your system, you can try running the MuRaL commands with the
mural_vx.x.x.sif file.
If your machine has GPUs and you want to use GPU resources for MuRaL
tools, please remember to set the --nv flag for Singularity commands.
See the following examples:
singularity exec --nv /path/to/mural_vx.x.x.sif mural_snv train ...
singularity exec --nv /path/to/mural_vx.x.x.sif mural_snv transfer ...
For prediction tasks, it is recommended to use only CPUs so that you can run many prediction tasks in parallel. See the example below:
singularity exec /path/to/mural_vx.x.x.sif ...
For more about Singularity, please refer to the Singularity documentation.
Tools and examples
Command Line Tools
All MuRaL tools are available via command line. For detailed usage,
run any command with the -h help option.
See subsequent sections for specific examples.
Main Commands
MuRaL-snv
|
Train new mutation rate models from scratch |
|
Find the best SNV rate model from trial results (requires ‘progress.csv’) |
|
Fine-tune models using transfer learning |
|
Predict mutation rate predictions |
|
Evaluate model performance metrics |
|
Calculate scaling factors |
|
Convert predictions to per-generation rates |
MuRaL-indel
|
Train new INDEL rate models |
|
Find the best INDEL rate model from trial results (requires ‘progress.csv’) |
|
Apply transfer learning for INDEL models |
|
Predict INDEL mutation rates |
|
Evaluate INDEL model performance |
|
Calculate scaling factors |
|
Scale INDEL rate predictions |
Model training
Both mural_snv train and mural_indel train commands train MuRaL models using
training and validation mutation data. Training results are saved in different locations
based on whether Ray is used for hyperparameter search(–use_ray):
Without Ray:
./results/folderWith Ray:
./ray_results/folder
Input Requirements
Required Files:
Reference sequence (FASTA format)
Training data (BED format)
Validation data (BED format, optional)
Note: If the validation data file isn’t provided, a fraction of the sites sampled from the training data file are used as validation data.
Input training and validation data files are in BED format (more info about BED format here). Some example lines of an input BED file are shown below.
chr1 2333436 2333437 . 0 +
chr1 2333446 2333447 . 2 -
chr1 2333468 2333469 . 1 -
chr1 2333510 2333511 . 3 -
chr1 2333812 2333813 . 0 -
In the BED-formatted lines above, the 5th column is used to represent
mutation status: usually, ‘0’ means the non-mutated status and other
numbers for specific mutation types (e.g. ‘1’ for ‘A>C’, ‘2’ for ‘A>G’,
‘3’ for ‘A>T’). You can specify an arbitrary order for a group of
mutation types with incremental numbers starting from 0, but make sure
that the same order is consistently used in training, validation and
testing datasets. Importantly, the training and validation BED file MUST
BE SORTED by chromosome coordinates. You can sort BED files by
bedtools sort or sort -k1,1 -k2,2n.
Recurrent mutation BED format
When analyzing recurrent mutations (sites mutated multiple times across
individuals), use the --recurrent flag during training, prediction
and evaluation. In this mode, the 4th column (name field) of the BED
file encodes per-site information in a semicolon-separated format:
chrom:start;ref>alt;AC;count, where AC is the allele count (integer;
-1 if unavailable) and the last field is the observed mutation count:
chr1 238329 238330 chr1:238329;G>A;-1;3 1 +
chr1 238331 238332 chr1:238331;G>A;-1;1 1 -
chr1 238335 238336 . 0 +
In the example above, the site at chr1:238329 has 3 independent mutation
events (count=3), while chr1:238331 has 1 event. Non-mutated sites use
. as the name field and are assigned a weight of 1.
During training with --recurrent, per-site counts are used as sample
weights in the cross-entropy loss, so sites with more independent
mutations contribute proportionally more to the gradient.
Output data
mural_snv train or mural_indel train saves the model information at each checkpoint,
normally at the end of each training epoch of a trial. The checkpointed
model files during training are saved under folders named like:
Non-Ray Execution (default):
./results/{experiment_name}/Train_{trial_id}/checkpoint_{epoch}/
├── model # Learned parameters
├── model.config.pkl # Model hyperparameters
└── model.fdiri_cal.pkl # Calibration model (if validation data provided)
Ray Execution:
./ray_results/{experiment_name}/Train_{trial_id}/checkpoint_{epoch}/
├── model
├── model.config.pkl
└── model.fdiri_cal.pkl
In the above folder, the ‘model’ file contains the learned model
parameters. The ‘model.config.pkl’ file contains configured
hyperparameters of the model. The ‘model.fdiri_cal.pkl’ file (if
exists) contains the calibration model learned with validation data,
which can be used for calibrating predicted mutation rates. These
files can be used in downstream analyses such as model prediction and
transfer learning. The ‘progress.csv’ files in ‘Train_xxx’ folders
contain important information for each training epoch of trials
(e.g., validation loss, used time, etc.).
You can use the command mural_snv get_best_model or
mural_indel get_best_model to find the best model per trial after
training.
# For SNV: running trials without Ray (default)
mural_snv get_best_model ./results/your_experiment_name
# For SNV: running trials using Ray
mural_snv get_best_model ./ray_results/your_experiment_name
# For INDEL: running trials without Ray (default)
mural_indel get_best_model ./results/your_experiment_name
# For INDEL: running trials using Ray
mural_indel get_best_model ./ray_results/your_experiment_name
The output lists all trials sorted by validation loss (ascending). Use the top result for predictions. Example:
./results/your_experiment_name/Train_zoq97_00000/checkpoint_8 0.401637 # ← Best model (lowest loss)
./results/your_experiment_name/Train_zoq97_00002/checkpoint_8 0.407693
./results/your_experiment_name/Train_zoq97_00001/checkpoint_6 0.445094
Example 1: Basic Training
MuRaL-snv
The following commands will train a SNV model by running two trials (default:--n_trials 2),
using data in ‘snv/data/training.sorted.bed’ for training. The training
results will be saved under the folder ‘./results/example1/’ or ‘./ray_results/example1/’.
Default values will be used for other unspecified arguments. Note
that, by default, 10% of the sites sampled from ‘training.sorted.bed’
is used as validation data (i.e. --valid_ratio 0.1). You can run
this example under the ‘examples/’ folder in the package.
# serially running two trials (default)
mural_snv train --ref_genome snv/data/seq.fa --train_data snv/data/training.sorted.bed \
--experiment_name example1 > test1.out 2> test1.err
# parallelly running two trials using Ray, with 3 CPUs each trial (6 requested CPUs in total)
mural_snv train --ref_genome snv/data/seq.fa --train_data snv/data/training.sorted.bed \
--use_ray --ray_ncpus 6 --cpu_per_trial 3 --experiment_name example1 \
> test1.out 2> test1.err
Note
If your machine has sufficient resources to execute multiple trials in parallel,
it is recommended to add the --use_ray option. Using Ray allows for better resource
scheduling. If running multiple trials serially or running only a single trial (set --n_trials 1),
it is recommended not to use --use_ray, which can improve the runtime speed by approximately
2-3 times for each trial.
MuaRaL-indel
The following commands train an INDEL model with two trials (default: --n_trials 2),
using indel/data/training.sorted.bed as input. Parameters are identical to SNV training,
but with two key differences:
Uses larger default context window (
--distal_radius 4000) to improve INDEL prediction accuracy while maintaining reasonable computational requirements
# serially running two trials (default)
mural_indel train --ref_genome indel/data/seq.fa \
--train_data indel/data/training.sorted.bed \
--experiment_name example1 > test1.out 2> test1.err
# parallelly running two trials using Ray, with 3 CPUs each trial (6 requested CPUs in total)
mural_indel train --ref_genome indel/data/seq.fa \
--train_data indel/data/training.sorted.bed \
--use_ray --ray_ncpus 6 --cpu_per_trial 3 --experiment_name example1 \
--experiment_name example1 > test1.out 2> test1.err
Note
–use_reverse should be added, if insertion model is trained.
Example 2: Training with large sequence
MuRaL-snv
The following command will use data in ‘snv/data/training.sorted.bed’
as training data and a separate ‘snv/data/validation.sorted.bed’ as
validation data. The option --local_radius 7 means that length of
the local sequence used for training is 7*2+1 = 15 bp.
--distal_radius 200 means that length of the expanded sequence
used for training is 200*2+1 = 401 bp. You can run this example
under the ‘examples/’ folder in the package.
mural_snv train --ref_genome snv/data/seq.fa \
--train_data snv/data/training.sorted.bed \
--validation_data snv/data/validation.sorted.bed \
--n_trials 2 --local_radius 7 \
--distal_radius 200 --experiment_name example2 \
> test2.out 2> test2.err
MuRaL-indel
The following command will use data in ‘indel/data/training.sorted.bed’
as training data and a separate ‘indel/data/validation.sorted.bed’ as
validation data. --distal_radius 4000 means that length of the expanded sequence
used for training is 4000*2 = 8000 bp. You can run this example
under the ‘examples/’ folder in the package.
# For indel, 4k radius is recommend
mural_indel train --ref_genome indel/data/seq.fa \
--train_data indel/data/training.sorted.bed \
--validation_data indel/data/validation.sorted.bed \
--n_trials 2 \
--distal_radius 4000 --experiment_name example2 \
> test2.out 2> test2.err
Example 3: Training acceleration
For large sequence contexts (distal_radius > 1000), data loading may become a bottleneck during training.
To mitigate this, specify additional CPUs per trial using --cpu_per_trial.
The following example demonstrates using 3 extra CPUs to accelerate data loading(applies equally to both mural_snv and mural_indel):
mural_snv train --ref_genome snv/data/seq.fa \
--train_data snv/data/training.sorted.bed \
--cpu_per_trial 4 \
--experiment_name example3 > test3.out 2> test3.err
mural_indel train --ref_genome indel/data/seq.fa \
--train_data indel/data/training.sorted.bed \
--cpu_per_trial 4 \
--experiment_name example3 > test3.out 2> test3.err
Example 4: Training with limited resources
If RAM memory or GPU memory limits the usage of mural_snv train or mural_indel train (which may happen with large
expanded sequences used for training), please refer to following suggestions.
Since v1.1.2, we split the genomes into segments of a spefic size to facilitate data preprocessing (see illustration in the panel A of the figure below).
To reduce RAM memory, you can set smaller values for --segment_center and --sampled_segments.
The default value of --segment_center is 300,000 bp, meaning maximum encoding unit of the
sequence is 300000+2*distal_radius bp (see illustration in the panel A of the figure below). It is the key parameter
for trade-off between RAM memory usage and data preprocessing speed. You can reduce this
(e.g., 50,000 bp) at the cost of an acceptable loss in data preprocessing speed.
The second option is to set smaller value for --sampled_segments (default 10). If changing this, you should also
check the performance of trained model, because --sampled_segments may also influence model performance
sometimes. The impact of the two parameters on RAM usage can be visualized in the following figure (panels B & C):

To reduce GPU memory, it is recommended to reduce --batch_size (default 128). You can set smaller values (e.g., 64, 32, 16, etc.).
In addition, you may also consider using smaller models (e.g., reducing --distal_radius, --CNN_out_channels, etc.), but that may affect model performance.
You can run the following commands under the ‘examples/’ folder in the package.
MuRaL-snv
# use less RAM memory
mural_snv train --ref_genome snv/data/seq.fa \
--train_data snv/data/training.sorted.bed \
--validation_data snv/data/validation.sorted.bed \
--n_trials 2 --local_radius 7 \
--distal_radius 6400 --segment_center 100000 \
--sampled_segments 4 --experiment_name example4 \
> test4.out 2> test4.err
# use less GPU memory
mural_snv train --ref_genome snv/data/seq.fa \
--train_data snv/data/training.sorted.bed \
--validation_data snv/data/validation.sorted.bed \
--n_trials 2 --local_radius 7 \
--batch_size 64 \
--distal_radius 6400 --experiment_name example4 \
> test4.out 2> test4.err
MuRaL-indel
# use less RAM memory
mural_indel train --ref_genome indel/data/seq.fa \
--train_data indel/data/training.sorted.bed \
--validation_data indel/data/validation.sorted.bed \
--n_trials 2 --local_radius 7 \
--distal_radius 6400 --segment_center 100000 \
--sampled_segments 4 --experiment_name example4 \
> test4.out 2> test4.err
# use less GPU memory
mural_indel train --ref_genome indel/data/seq.fa \
--train_data indel/data/training.sorted.bed \
--validation_data indel/data/validation.sorted.bed \
--n_trials 2 --local_radius 7 \
--batch_size 64
--distal_radius 6400 --experiment_name example4 \
> test4.out 2> test4.err
Note
The RAM memory usage can be estimated as:
sampled_segments * segment_center * 4 * (2 * distal_radius + 1) * 4 / 2^30 + C (GB)
where:
Cis a constant term ranging between 5 and 12 GB;seq_lengthis (2 * distal_radius + 1) for SNV,seq_lengthis (2 * distal_radius) for INDEL,
When experiencing insufficient RAM, this formula helps determine appropriate parameter values.
Example 4b: Training with recurrent mutations
When training on recurrent mutation data (where the same genomic site may
be mutated independently in multiple individuals), add the --recurrent
flag. This uses per-site mutation counts as sample weights in the loss
function, ensuring that sites with more independent mutation events
contribute proportionally to training.
mural_snv train --ref_genome snv/data/seq.fa \
--train_data snv/data/recurrent_training.sorted.bed \
--validation_data snv/data/recurrent_validation.sorted.bed \
--recurrent --n_trials 2 --experiment_name example4b \
> test4b.out 2> test4b.err
Note
The recurrent mutation BED files in the example directory are synthetic
data generated from the standard training/validation files via
generate_recurrent_data.py. They are for demonstration purposes only.
Model prediction
mural_snv predict and mural_indel predict predicts mutation rates for all sites in a BED file
based on a trained model.
Input Requirements
Reference genome (FASTA format)
Input BED file (formatted identically to training BED files) - Column 5 can be set to ‘0’ if mutation status is unknown
Trained model files(generated by
mural_snv trainormural_indel train): -model(learned parameters) -model.config.pkl(model configuration)Optional calibration file: -
model.fdiri\_cal.pkl(for calibrating predicted mutation rates)
Output data
The output of mural_snv predict is a tab-separated file containing
the sequence coordinates (BED-formatted) and the predicted probabilities
for all possible mutation types. Usually, the ‘prob0’ column contains
probabilities for the non-mutated class and other ‘probX’ columns for
mutated classes. Some example lines of a prediction output file are shown below.
SNV output:
chrom start end strand mut_type prob0 prob1 prob2 prob3
chr1 10006 10007 - 0 0.9797 0.003134 0.01444 0.002724
chr1 10007 10008 + 0 0.9849 0.005517 0.00707 0.002520
chr1 10008 10009 + 0 0.9817 0.004801 0.01006 0.003399
chr1 10012 10013 - 0 0.9711 0.004898 0.02029 0.003746
INDEL output:
chrom start end strand mut_type prob0 prob1 prob2 ... prob7
chr1 10006 10007 - 0 0.9649 0.008623 0.00680 ... 0.012542
chr1 10007 10008 + 0 0.9849 0.005517 0.00707 ... 0.009099
chr1 10008 10009 + 0 0.9717 0.007114 0.00385 ... 0.008034
chr1 10012 10013 - 0 0.9706 0.010121 0.00476 ... 0.006360
Example 5: Mutation Rate Prediction
MuRaL-snv
Predicts mutation rates for sites in ‘snv/data/testing.bed.gz’ using model from ‘snv/models/checkpoint_6/’:
mural_snv predict --ref_genome snv/data/seq.fa --test_data snv/data/testing.bed.gz \
--model_path snv/models/checkpoint_6/model \
--model_config_path snv/models/checkpoint_6/model.config.pkl \
--calibrator_path snv/models/checkpoint_6/model.fdiri_cal.pkl \
--pred_file testing.ckpt6.fdiri.tsv.gz \
--cpu_only > test5.out 2> test5.err
For recurrent mutation data, add --recurrent so that correlation
calculations use observed mutation counts instead of binary
presence/absence:
mural_snv predict --ref_genome snv/data/seq.fa --test_data snv/data/recurrent_validation.sorted.bed \
--model_path snv/models/checkpoint_6/model \
--model_config_path snv/models/checkpoint_6/model.config.pkl \
--calibrator_path snv/models/checkpoint_6/model.fdiri_cal.pkl \
--pred_file testing.ckpt6.fdiri.tsv.gz \
--recurrent --cpu_only > test5.out 2> test5.err
MuRaL-indel
mural_indel predict --ref_genome indel/data/seq.fa --test_data indel/data/testing.bed.gz \
--model_path indel/models/checkpoint_9/model \
--model_config_path indel/models/checkpoint_9/model.config.pkl \
--calibrator_path indel/models/checkpoint_9/model.fdiri_cal.pkl \
--pred_file testing.ckpt9.fdiri.tsv.gz \
--cpu_only > test5.out 2> test5.err
Transfer learning
mural_snv transfer and mural_indel transfer trains MuRaL models like
mural_snv train and mural_indel train but initializes model parameters
with learned weights from a pre-trained model.
Its training results are also saved under the ‘./results/’ or ‘./ray_results/’
folder.
Input Requirements
Required files:
Reference genome (FASTA format)
Training data (BED format)
Pre-trained model files:
model(learned parameters)model.config.pkl(model configuration)
Optional:
Validation data (BED format)
Calibration model (
model.fdiri_cal.pkl)
Output data
Output data has the same structure as that of mural train.
Example 6: transfer learning commands
MuRaL-snv
The following command will train a transfer learning SNV model using training data in ‘snv/data/training_TL.sorted.bed’, the validation data in ‘snv/data/validation.sorted.bed’, and the model files under ‘models/checkpoint_6/’. You can run this example under the ‘examples/’ folder in the package.
mural_snv transfer --ref_genome snv/data/seq.fa \
--train_data snv/data/training_TL.sorted.bed \
--validation_data snv/data/validation.sorted.bed \
--model_path snv/models/checkpoint_6/model \
--model_config_path snv/models/checkpoint_6/model.config.pkl \
--train_all --init_fc_with_pretrained \
--experiment_name example6 > test6.out 2> test6.err
MuRaL-indel
The following command will train a transfer learning INDEL model using training data in ‘indel/data/training_TL.sorted.bed’, the validation data in ‘indel/data/validation.sorted.bed’, and the model files under ‘indel/models/checkpoint_6/’. You can run this example under the ‘examples/’ folder in the package.
mural_indel transfer --ref_genome indel/data/seq.fa \
--train_data indel/data/training_TL.sorted.bed \
--validation_data indel/data/validation.sorted.bed \
--model_path indel/models/checkpoint_9/model \
--model_config_path indel/models/checkpoint_9/model.config.pkl \
--train_all --init_fc_with_pretrained \
--experiment_name example6 > test6.out 2> test6.err
Calculating k-mer and regional correlations for evaluation
For model evaluation, since it is impossible to evaluate the accuracy
of predicted mutation rates at the single-nucleotide level, we employ
two metrics, k-mer correlation and regional correlation, to evaluate
model performance at the higher (summarized) levels. More details about
the two metrics can be found in the MuRaL paper. The k-mer and regional
correlations can be calculated with the predicted tsv files generated
by mural_snv predict and mural_indel predict.
The command mural_snv evaluate and mural_indel evaluate
is used for calculating k-mer and regional correlations.By default,
they output results for both k-mer correlation and regional correlation.
K-mer correlation analysis
Input data
The inputs for k-mer correlation analysis include the reference FASTA file, a prediction tsv file and the length of k-mer. Note that for evaluation, we need to provide a specific set of observed mutations (e.g. all available rare variants), which are stored in the 5th column of the prediction tsv files. These observed mutations are used for calculating observed mutation rates. We can change the content in the 5th column to evaluate model performance in different observed mutation data.
Output data
The outputs include a file (’*-mer.mut_rates.tsv’) storing predicted and observed k-mer rates of all possible mutation subtypes, and a file (’*-mer.corr.txt’) storing the k-mer correlations (Pearson’s r and p-value) of three mutation types in a specific order (e.g., for A/T sites, prob1, prob2 and prob3 are for A>C, A>G and A>T, respectively).
# example of '*-mer.mut_rates.tsv'
type avg_obs_rate1 avg_obs_rate2 avg_obs_rate3 avg_pred_prob1 avg_pred_prob2 avg_pred_prob3 number_of_mut1 number_of_mut2 number_of_mut3 number_of_all
TAG 0.006806776385512125 0.010141979926438501 0.012039461380213204 0.012744358544122413 0.01817057941563919 0.021860978496512425 3494 5206 6180 513312
TAA 0.007517292690907348 0.011278023120833133 0.01318808653952362 0.013600087566977897 0.019697007577734515 0.024266536859123104 7214 10823 12656 959654
AAA 0.0068964404639771226 0.010705555691654661 0.009617493130148654 0.012599749576515839 0.020442895433664586 0.01646869397956817 11542 17917 16096 1673617
# example of '*-mer.corr.txt'
3-mer prob1 0.9569216831654604 6.585788162834682e-09 # r and p for prob1
3-mer prob2 0.9326211281771537 1.4129640985193586e-07 # r and p for prob2
3-mer prob3 0.947146892265788 2.6848989196451608e-08 # r and p for prob3
Example 7 : k-mer correlation commands
MuRaL-snv
The following commands use the prediction file ‘testing.ckpt4.fdiri.tsv.gz’ to calculate 3-mer, 5-mer and 7-mer correlations:
mural_snv evaluate --pred_file testing.ckpt4.fdiri.tsv.gz --ref_genome snv/data/seq.fa --kmer_length 3 --kmer_only --out_prefix test_kmer_corr
mural_snv evaluate --pred_file testing.ckpt4.fdiri.tsv.gz --ref_genome snv/data/seq.fa --kmer_length 5 --kmer_only --out_prefix test_kmer_corr
mural_snv evaluate --pred_file testing.ckpt4.fdiri.tsv.gz --ref_genome snv/data/seq.fa --kmer_length 7 --kmer_only --out_prefix test_kmer_corr
For recurrent mutation data, add --recurrent so that observed rates
reflect the actual number of independent mutation events rather than
binary presence/absence:
mural_snv evaluate --pred_file testing.ckpt4.fdiri.tsv.gz --ref_genome snv/data/seq.fa --kmer_length 3 --kmer_only --recurrent --out_prefix test_kmer_corr
MuRaL-indel
The following commands use the prediction file ‘testing.ckpt4.fdiri.tsv.gz’ to calculate 2-mer, 4-mer and 6-mer correlations:
mural_indel evaluate --pred_file testing.ckpt9.fdiri.tsv.gz --ref_genome indel/data/seq.fa --kmer_length 2 --kmer_only --out_prefix test_kmer_corr
mural_indel evaluate --pred_file testing.ckpt9.fdiri.tsv.gz --ref_genome indel/data/seq.fa --kmer_length 4 --kmer_only --out_prefix test_kmer_corr
mural_indel evaluate --pred_file testing.ckpt9.fdiri.tsv.gz --ref_genome indel/data/seq.fa --kmer_length 6 --kmer_only --out_prefix test_kmer_corr
Regional correlation analysis
Input data
The inputs for regional correlation analysis include a prediction tsv file and the window size. Like the k-mer correlation analysis, we need to provide a specific set of observed mutations in the 5th column of the prediction tsv files. These observed mutations are used for calculating observed regional mutation rates.
Output data
There are multiple output files. The files storing regional rates (’*.regional_rates.tsv’) have seven columns: chromosome name, the end position of the window, number of valid sites in the window, number of observed mutations in the window, average observed mutation rate, average predicted mutation rate in the window and the ‘used_or_deprecated’ label. The windows labeled ‘deprecated’ are not used in correlation analysis due to too few valid sites. The regional correlation (Pearson’s r and p-value) of the considered mutation type is given in the ‘*.corr.txt’.
# example of '*.regional_rates.tsv'
chrom end sites_count mut_type_total mut_type_avg avg_pred used_or_deprecated
chr3 100000 61492 576 0.009367072139465296 0.020374342255903233 used
chr3 200000 60680 531 0.008750823994726434 0.02025859070533955 used
chr3 300000 59005 499 0.00845691043131938 0.01882644280993153 used
...
# example of '*.corr.txt'
100Kb prob3 0.4999 6.040983e-16
Example 8: regional correlation commands
MuRaL-snv
The following command will calculate the regional correlation for 100Kb windows.
mural_snv evaluate --pred_file testing.ckpt4.fdiri.tsv.gz --window_size 100000 --regional_only --out_prefix test_region_corr
With --recurrent, observed counts reflect the number of independent
mutation events per window:
mural_snv evaluate --pred_file testing.ckpt4.fdiri.tsv.gz --window_size 100000 --regional_only --recurrent --out_prefix test_region_corr
MuRaL-indel
The following command will calculate the regional correlation for 100Kb windows.
mural_indel evaluate --pred_file testing.ckpt9.fdiri.tsv.gz --window_size 100000 --regional_only --out_prefix test_region_corr
Visualization of correlation results
You can run the commands like below to extract k-mer correlations and corresponding p-values for further visualization:
# For SNV
cat test.{3,5,7}-mer.corr.txt | awk 'BEGIN{print "k-mer\tmut_type\tcorrelation\tp-value"}{print;}' > kmer_correlations.tsv
# For INDEl
cat test.{2,4,6}-mer.corr.txt | awk 'BEGIN{print "k-mer\tmut_type\tcorrelation\tp-value"}{print;}' > kmer_correlations.tsv
The resulting ‘kmer_correlations.tsv’ file is tab-delimited, looking like:
# For SNV
k-mer mut_type correlation p-value
3-mer A>C 0.8527 2.7049e-05
3-mer A>G 0.8453 3.7235e-05
...
or
# For INDEL
kmer mut_type correlation p-value
2-mer A>C 0.8527 2.7049e-05
2-mer A>G 0.8453 3.7235e-05
...
The following python code can be used for generating bar plots for k-mer correlations:
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
df = pd.read_table('kmer_correlations.tsv')
plt.figure(figsize=(6,4))
sns.catplot(x="mut_type", y="correlation", kind="bar", hue="k-mer", data=df, palette="Blues_r")
plt.title('Bar plots of k-mer correlations')
plt.savefig('kmer_correlations.jpg', bbox_inches='tight')
The plot looks like below:

Similarly, one can generate bar plots for regional correlations for evaluation.
In addition, based on the output of calc_region_corr above, we can
visualize how predicted rates fit observed rates for windows across
a chromosome or a specific region. First, we should standardize the
observed rates and the predicted rates for all windows by using z-score
transformation. Then we select some regions to generate the plots. Below
we use the results for 100Kb windows and A>G mutation type, and the region
selected is from 15Mb to 23.6Mb. The solid line indicates average
predicted mutation rates and the shade for average observed mutation
rates:
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from sklearn import preprocessing
from scipy.stats import pearsonr
df = pd.read_table('test2.100Kb.prob2.regional_rates.tsv')
df = df[df['used_or_deprecated'] == 'used']
#z-score preprocessing
avg_obs = preprocessing.scale(df['avg_obs'])
avg_pred = preprocessing.scale(df['avg_pred'])
data = {'window_end':df['window_end'],'avg_obs':list(avg_obs),'avg_pred':list(avg_pred)}
df1 = pd.DataFrame(data)
#select the region
df2 = df1[143:229]
corr = pearsonr(df2['avg_obs'],df2['avg_pred'])
print("Correlation of the selected regions is %f, p-value is %f" %(corr[0],corr[1]))
#plot
fig, ax = plt.subplots(1, figsize=(10, 2))
ax.set_xlabel("Chr3(Mb)")
ax.fill_between(df2['window_end']/1000000,df2['avg_obs'], alpha=0.3, color = 'Grey')
ax.plot(df2['window_end']/1000000,df2['avg_pred'], label="avg_pred", linewidth = 1.5)
plt.ylabel('average mutation rate (Z-score)')
plt.savefig('regional_rates.jpg', bbox_inches = 'tight')
The plot looks like below:

Scaling MuRaL-predicted mutation rates to per base per generation rates
The raw MuRaL-predicted mutation rates are not mutation rates per bp per generation. To obtain a mutation rate per bp per generation for each nucleotide, one can scale the MuRaL-predicted rates using reported genome-wide de novo mutation rate and spectrum per generation.
Calculate scaling factors: Use the appropriate command for your mutation type:
mural_snv calc_scaling_factor- for SNV site groups (A/T, C/G, CpG sites)mural_indel calc_scaling_factor- for INDEL site types (insertions, deletion starts, deletion ends)
Apply scaling: use the command
mural_snv scaleormural_indel scaleto scale mutation rates in prediction files via scaling factors
Note
Cannot directly compare raw rates from different MuRaL models
Genome-wide rate accuracy affects between-species comparison
Scaling does not affect k-mer/regional correlation calculations
Example 9 : Scaling workflow
MuRaL-snv
Step 1: Calculate scaling factor
Here is an example for scaling mutation rates for A/T sites. Suppose that we have the following proportions of different mutation types and proportions of different site groups in a genome. In addition, suppose we already know from previous research that the genome-wide mutation rate per generation of the species is 5e−9 per base per generation. If the per generation mutation rate is not available for the studied species, one may use the estimate from a closely related species.
#mutation_type proportion
AT_mutations 0.355
nonCpG_mutations 0.423
CpG_mutations 0.222
#site_group proportion
AT_sites 0.475
nonCpG_sites 0.391
CpG_sites 0.134
To calculate the scaling factor, we need to have the predicted mutation rates for a set of representative sites based on a trained model. It is recommended to use the validation sites at the training step whose size is relatively small and representative enough. For instance, the following command is for obtaining predicted mutation rates for validation sites of the A/T model.
mural_snv predict --ref_genome snv/data/seq.fa --test_data snv/data/AT_validation.sorted.bed \
--model_path models/checkpoint_6/model --model_config_path models/checkpoint_6/model.config.pkl \
--calibrator_path models/checkpoint_6/model.fdiri_cal.pkl \
--pred_file AT_validation.ckpt6.fdiri.tsv.gz \
--cpu_only > test.out 2> test.err
Next, the command mural_snv calc_snv_scaling_factor will be used to compute the scaling
factor based on the predicted rates, the proportions of A/T mutation types and
proportions of A/T sites in the genome, and the genome-wide per generation mutation
rate.
mural_snv calc_scaling_factor --pred_files AT_validation.ckpt6.fdiri.tsv.gz --genomewide_mu 5e-9
--m_proportions 0.355 --g_proportions 0.475 > scaling_factor.out
Output file ‘scaling_factor.out’ may look like the following:
pred_file: AT_validation.ckpt6.fdiri.tsv.gz
genomewide_mu: 5e-09
n_sites: 84000
g_proportion: 0.475
m_proportion: 0.355
prob_sum: 4.000e+03
scaling factor: 7.848e-08
Step 2: Apply scaling
Finally, the obtained scaling factor 7.848e-08 is used to scale all the
predicted rates of all A/T sites using mural_snv scale --scale_factors. You can run
separately for each chromosome.
mural_snv scale --pred_file AT_chr1.tsv.gz --scale_factor 7.848e-08 --out_file AT_chr1.scaled.tsv.gz
Similarly, you can generate the scaled mutation rates for non-CpG and CpG sites like the above example. More details can be found in the MuRaL paper.
MuRaL-indel
Step 1: Calculate scaling factor
Here is an example for scaling mutation rates for insertion sites. Suppose that we have the following proportions of different INDEL mutation types and proportions of different site groups in a genome. In addition, suppose we already know from previous research that the genome-wide mutation rate per generation of the species is 1.5e−9 per base per generation. If the per generation mutation rate is not available for the studied species, one may use the estimate from a closely related species.
#INDEL proportion
insertion 0.352
deletion start 0.648
deletion end 0.648
Note: Each deletion has two breakpoints—designated as the deletion start and deletion end. These two labels refer to the same deleted interval, so, same proportions are used for both.
To calculate the scaling factor, we need to have the predicted mutation rates for a set of representative sites based on a trained model. It is recommended to use the validation sites at the training step whose size is relatively small and representative enough. For instance, the following command is for obtaining predicted mutation rates for validation sites of the INDEL model.
mural_indel predict --ref_genome indel/data/seq.fa --test_data indel/data/insertion_validation.sorted.bed \
--model_path models/checkpoint_6/model --model_config_path models/checkpoint_6/model.config.pkl \
--calibrator_path models/checkpoint_6/model.fdiri_cal.pkl \
--pred_file insertion_validation.ckpt6.fdiri.tsv.gz \
--cpu_only > test.out 2> test.err
Next, the command mural_indel scale will be used to compute the scaling
factor based on the predicted rates, the proportions of insertion mutation types,
and the genome-wide per generation mutation rate.
mural_indel calc_scaling_factor --pred_files insertion_validation.ckpt6.fdiri.tsv.gz --genomewide_mu 1.5e-9
--m_proportions 0.352 > scaling_factor.out
Output file ‘scaling_factor.out’ may look like the following:
pred_file: insertion_validation.ckpt6.fdiri.tsv.gz
genomewide_mu: 1.5e-09
n_sites: 221240912
m_proportion: 0.352
prob_sum: 4.000e+03
scaling factor: 6.271e-09
Step 2: Apply scaling
Finally, the obtained scaling factor 6.271e-09 is used to scale all the
predicted rates of all insertion sites using mural_indel scale --scale_factors. You can run
separately for each chromosome.
mural_indel scale --pred_file insertion_chr1.tsv.gz --scale_factor 6.271e-09 --out_file insertion_chr1.scaled.tsv.gz
Similarly, you can generate the scaled mutation rates for deletion start and deletion end sites like the above example. More details can be found in the MuRaL-indel paper.
Trained models and predicted mutation rate maps of multiple species
MuRaL-snv
Trained models (by MuRaL v1.0.0) for four species - Homo sapiens, Macaca mulatta, Arabidopsis thaliana and Drosophila melanogaster are provided in the ‘models/*/SNV’ folder of the package. One can use these model files for prediction or transfer learning.
Predicted single-nucleotide mutation rate maps (by MuRaL v1.0.0) for these genomes are available at ScienceDB.
MuRaL-indel
Trained models (by MuRaL v1.2.0) for four species - Homo sapiens, Macaca mulatta, Arabidopsis thaliana and Drosophila melanogaster are provided in the ‘models/*/INDEL’ folder of the package. One can use these model files for prediction or transfer learning.
Predicted base-resolution INDEL mutation rate maps (by MuRaL v1.2.0) for these genomes are available at ScienceDB.
Citation
Fang Y, Deng S, Li C. A generalizable deep learning framework for inferring fine-scale germline mutation rate maps. Nature Machine Intelligence (2022) doi:10.1038/s42256-022-00574-5
Deng S, Song H, Li C. A deep learning framework for building INDEL mutation rate maps. Briefings in Bioinformatics (2025) doi:10.1093/bib/bbag212 <https://doi.org/10.1093/bib/bbag212>
Contact
For reporting issues or requests related to the package, please use GitHub Issues or write to mural-project@outlook.com.